Nel gennaio 2017 un uomo di 45 anni con anamnesi patologica sostanzialmente negativa inizia a manifestare diversi sintomi, tra cui un’astenia sempre più marcata associata a dispnea per sforzi lievi, tosse stizzosa, lipotimie, difficoltà digestive e un progressivo calo ponderale con perdita di circa 10 kg nel corso dell’anno. Inizia qui il suo iter tra ospedali e visite specialistiche, senza arrivare a una diagnosi. Infine una visita cardiologica sul territorio evidenzia all’ecocardiogramma un fenotipo ipertrofico, motivo per cui il paziente viene indirizzato all’ Ospedale S. Orsola Malpighi di Bologna, dove è ricoverato nel gennaio 2018.
All’esame obiettivo all’ingresso erano presenti minimi segni di scompenso cardiaco, quali turgore giugulare con pressione venosa centrale stimata pari a circa 10 cm H2O ed epatomegalia non dolente. I parametri vitali erano nella norma con tendenza all’ipotensione (PA: 90/60 mmHg).
L’ ECG evidenziava ritmo sinusale a 97 bpm, pseudonecrosi anteriore con alterazioni diffuse della ripolarizzazione ventricolare e bassi voltaggi periferici (Figura 1).
I principali esami laboratoristici risultavano nella norma.

Veniva ripetuto l’ecocardiogramma che mostrava un severo aumento degli spessori parietali (SIVtd: 22 mm, PPtd: 22 mm) con buona funzione contrattile globale (FE: 70%) ed ipercontrattilità apicale, ma riduzione della funzione longitudinale con onda S settale al TDI pari a 5 cm/s. Si evidenziavano inoltre un aspetto a granular sparkling del miocardio e un ispessimento delle valvole atrioventricolari e del setto interatriale, reperti suggestivi per malattia infiltrativa (Figura 2).

Per approfondire tale sospetto, venivano richiesti ulteriori esami laboratoristici che evidenziavano la presenza di catene leggere libere kappa all’immunofissazione sierica e urinaria con valori di catene kappa libere nel siero pari a 876.3 mg/l e rapporto kappa/lambda pari a 324.56. Inoltre si segnalava un importante aumento di NTproBNP pari a 2180 pg/ml e della troponina I pari a 125 ng/l.
Il sospetto di amiloidosi cardiaca AL diveniva quindi primario, e al fine di confermare e stadiare la malattia ematologica venivano eseguiti:
- biopsia osteo-midollare: cellularità 50%. Infiltrato plasmacellulare composto da elementi prevalentemente ad abito maturo, a distribuzione interstiziale ed in aggregati. All’immunoistochimica monotipia per catena leggera kappa delle immunoglobuline;
- PET total body con FDG: non aree di patologico iperaccumulo del radiofarmaco da riferire a lesioni ad elevata attività metabolica. In particolare non patologici iperaccumuli a livello osteo-midollare;
- biopsia del grasso periombelicale: positiva la ricerca di amiloide con colorazione rosso congo.
Per caratterizzare meglio il quadro emodinamico veniva eseguito un cateterismo cardiaco destro (Tabella 1) documentante un lieve aumento delle pressioni di riempimento biventricolari con ridotto indice cardiaco a riposo.
| Atrio destro | 9 mmHg |
| PAP s/d/m | 32/9/23 mmHg |
| CPWP | 18 mmHg |
| IC | 1.9 l/min/m2 |
| Resistenze arteriose polmonari | 2.62 UW |
Tabella 1. Cateterismo cardiaco destro documentante un lieve aumento delle pressioni di riempimento biventricolari e una riduzione dell’indice cardiaco a riposo.
A questo punto siamo di fronte alla diagnosi di amiloidosi cardiaca AL secondaria a mieloma plasmacellulare a basso grado; la prognosi del paziente risulta influenzata sia dalla malattia ematologica primitiva sia dalla localizzazione d’organo a livello cardiaco.
Il caso veniva quindi discusso collegialmente con un tema multidisciplinare che comprendeva cardiologi ed ematologi. La strategia terapeutica iniziale prevedeva:
- terapia medica dello scompenso cardiaco: sono stati utilizzati diuretici e risparmiatori del potassio in quanto gli ACE-inibitori non erano tollerati per ipotensione sintomatica e rapido deterioramento della funzionalità renale e i beta-bloccanti non sono indicati nei pazienti con amiloidosi per il noto rischio di blocchi del sistema di conduzione;
- trattamento chemioterapico con 4 cicli ciascuno costituito da 4 somministrazioni settimanali di bortezomib e desametasone in associazione alla talidomide;
- stretto monitoraggio clinico e strumentale, per cui il paziente è rimasto ricoverato in ambiente cardiologico per tutta la durata del trattamento.
Come messo in evidenza dall’andamento dei valori plasmatici di NT-proBNP e dei livelli di catene leggere kappa libere sieriche (Figura 3), al termine del quarto ciclo di chemioterapial paziente presentava remissione parziale della malattia ematologica e un labile compenso emodinamico, per cui nel corso del ricovero è stato necessario aumentare il dosaggio giornaliero di diuretico.
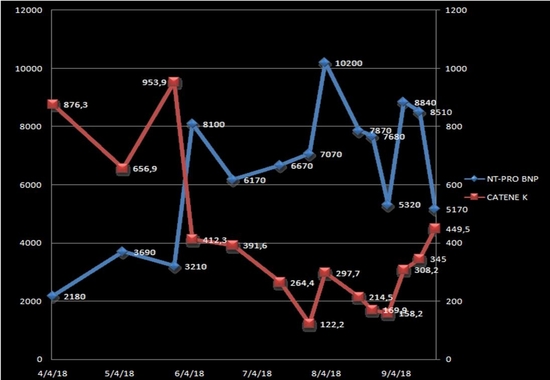
Il caso è stato quindi ridiscusso ed è stata stabilita una strategia terapeutica di doppio trapianto: trapianto ortotopico di cuore e trapianto autologo di cellule staminali ematopoietiche, da effettuarsi dopo aver concluso anche il quinto e sesto ciclo di chemioterapia con bortezomib, desametasone e talidomide.
Nel luglio 2018 veniva eseguita aferesi delle cellule staminali ematopoietiche.
Veniva quindi eseguito screening urgente per trapianto cardiaco che escludeva controindicazioni assolute e il paziente veniva inserito in lista urgente in considerazione della necessità di stabilizzare il quadro cardiologico prima di procedere al trapianto autologo di cellule staminali. Nell’ottobre 2018 il paziente veniva sottoposto a trapianto ortotopico di cuore. Lo studio anatomo-patologico del cuore espiantato documentava amiloidosi cardiaca AL con diffusi depositi interstiziali, vascolari, subendocardici e valvolari; erano inoltre visibili anche moderate alterazioni aspecifiche delle miocellule secondarie alla presenza dei depositi di amiloide (Figura 4).

Nel dicembre 2018 veniva eseguito trapianto autologo di cellule staminali ematopoietiche previo condizionamento mieloablativo.
Il paziente veniva quindi dimesso dopo 13 mesi di ricovero nel febbraio 2019, e a un mese dalla dimissione si presentava in buone condizioni generali, asintomatico dal punto di vista cardiovascolare, in classe NYHA I, in assenza di rigetto documentato alla BEM; dal punto di vista ematologico si notava una significativa riduzione delle catene kappa libere sieriche rispetto al pre-trapianto (catene kappa: 117 mg/l con rapporto kappa/lambda: 16.03).
Discussione
L’amiloidosi cardiaca è una patologia causata dall’accumulo di una proteina anomala all’interno delle strutture cardiache. Le due forme più comuni sono quella da transtiretina (TTR), wild type o mutata, e la forma da catene leggere delle immunoglobuline (AL). La prognosi dei pazienti con amiloidosi AL dipende dall’interessamento di un organo solido (in particolare il cuore, ma anche i reni o il fegato) e dalle dimensioni e dalla biologia del clone plasmacellulare (in particolare mieloma multiplo vs altre forme come plasmocitoma o MGUS) [1].
Secondo lo score descritto da Kumar et al., che prende in considerazione i livelli di NT-proBNP, di TnI e la differenza tra le catene leggere libere kappa e lambda descrivendo 4 stadi di malattia (0-1-2-3), il nostro paziente si trovava al terzo stadio di malattia, il più grave, con una sopravvivenza media attesa di circa 6 mesi [2].
I cardini della terapia nell’amiloidosi AL sono la chemioterapia e/o il trapianto di cellule staminali autologo, al fine di controllare la malattia ematologica di base, e l’eventuale trapianto di organo solido. Secondo le attuali indicazioni i pazienti non risultano candidabili al trapianto di cellule staminali quando vi è un preponderante interessamento cardiaco con elevazione della troponina; in questi casi risulta invece indicata una chemioterapia sistemica, per cui sono però da considerare i rischi legati alla cardiotossicità [3]. Poche sono le evidenze in questo campo, e in letteratura esistono solo piccole case series che descrivono il trattamento combinato con trapianto di cuore e successivo trapianto autologo di cellule staminali ematopoietiche; in tali report criteri di esclusione erano rappresentati dall’interessamento di un secondo organo solido e dalla malignità ematologica (mieloma multiplo) [4].
Conclusione
La cardiomiopatia amiloidotica AL è una condizione rara e di difficile diagnosi, determina una prognosi sfavorevole per i pazienti ed offre poche opzioni terapeutiche con scarse evidenze in letteratura.
Il nostro paziente si colloca all’esterno di qualunque linea guida e ha delineato una sfida terapeutica nella quale la collaborazione delle équipe cardiologica, cardiochirurgica ed ematologica ha rappresentato la chiave di volta per la cura.
Bibliografia
- Dispenzieri A et al., Treatment of Immunoglobulin Light Chain Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Statement. Mayo Clin Proc. n August 2015;90(8):1054-1081.
- Kumar S. et al., Revised Prognostic Staging System for Light Chain Amyloidosis Incorporating Cardiac Biomarkers and Serum Free Light Chain Measurements. J Clin Oncol. 2012 Mar 20; 30(9): 989–995.
- Morie A. Gertz., Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2018. Gertz Blood Cancer Journal (2018)8:44.
- Gillmore JD, Goodman HJ, Lachmann HJ, et al., Sequential heart and autologous stem cell transplantation for systemic AL amyloidosis. Blood. 2006;107:1227–1229.