
“Io, come Dio, non gioco ai dadi e non credo nelle coincidenze”
La cardiomiopatia ipertrofica (HCM) è una patologia prevalentemente a trasmissione autosomica dominante associata ad un aumento della morbilità e della mortalità. Essa è caratterizzata da un aumento della massa cardiaca, come conseguenza dell’ipertrofia e della fibrosi dei cardiomiociti, ed è causata principalmente da varianti genetiche dei geni che codificano per le proteine sarcomeriche. Tuttavia, dal 5% al 10% dei casi di HCM sono causati da difetti genetici rari, non correlati al sarcomero, comprese malattie neuromuscolari e metaboliche ereditarie, come la sindrome PRKAG2 (PS).
La sindrome PRKAG2 (PS) è causata da varianti genetiche del gene PRKAG2, che codifica per la subunità regolatoria della proteina chinasi gamma 2 attivata dall’adenosina monofosfato.
Nel cuore le varianti PRKAG2 determinano un accumulo di glicogeno all’interno dei cardiomiociti, responsabile di un quadro di Glicogenosi cardiaca non lisosomiale.
Essa è una malattia rara, con esordio clinico nella tarda adolescenza o nella terza decade di vita a trasmissione autosomica dominante (26 varianti genetiche), a penetranza incompleta e ad espressività clinica variabile nell’ambito della stessa famiglia.
Classicamente a tale patologia si associa una triade tipica:
1) Cardiomiopatia a fenotipo ipertrofico (da quadri lievi a quadri severi);
2) Preeccitazione ventricolare;
3) Disturbi della conduzione (Fibrillazione Atriale, Malattia del Nodo del Seno, Blocco atrio-ventricolare, blocchi di branca, Bradicardia sinusale, ecc,…).
A causa del complesso impatto elettrofisiologico della malattia, è stata suggerita un’incidenza di morte cardiaca improvvisa (SCD) prematura (<40 anni) fino al 20%.
La reale prevalenza della PS non è nota e i dati riguardanti le caratteristiche cliniche e gli esiti dei pazienti con PS sono scarsi, motivo per cui imbattersi nella pratica clinica in casi di pazienti affetti risulta spesso fonte di perplessità per il clinico.
Nel nostro caso familiare di sindrome PRKAG2 (PS), giungere al corretto inquadramento diagnostico e terapeutico è stato possibile a partire da un membro di tale famiglia, ovvero il paziente (A2), adulto di 41 anni di sesso maschile con evidenza elettrocardiografica di Preeccitazione Ventricolare (Figura 1) e verosimile ipertrofia ventricolare sinistra, che si è recato presso i nostri ambulatori di Aritmologia per il riscontro di episodi di Fibrillazione Atriale Parossistica (FAP) in giovane età.

Gli episodi di Fibrillazione Atriale sono stati cardiovertiti uno con Idrochinidina (antiaritmico di classe IA) e l’ultimo, avvenuto nel Giugno 2021 presso la nostra UOC, tramite cardioversione elettrica (CVE) alcune ore dopo infusione di Flecainide (antiaritmico di classe I C).
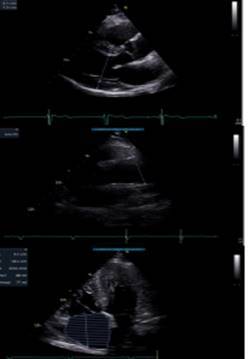
| Ecocardiogramma del paziente A2: Rimodellamento concentrico del ventricolo sinistro (SIV: 15 mm; DTD: 45 mm; PP: 10 mm). Eucinesia segmentaria con normale funzione globale di pompa (FE 60 %) e ridotta funzione sistolica longitudinale (S’ medio: 8.5 cm/s). Ventricolo destro di normali dimensioni e funzione sistolica longitudinale (TAPSE: 20 mm). Atrio sinistro di normali dimensioni (LAA: 13 cmq); dilatazione atriale destra (RAA: 23 cmq). Lieve lassità del setto interatriale. Radice aortica (36 mm) di normali dimensioni con normale movimento di apertura delle semilunari; al Doppler: normale flussimetria trans-valvolare. Aorta ascendente ectasica (44 mm). Valvola mitrale con normale movimento dei lembi; al Doppler normale flussimetria trans-valvolare. Normale funzione diastolica. Minimo rigurito tricuspidalico con normale stima indiretta della PAPs (20+5 mmHg). Non versamento pericardico. Vena cava inferiori di normali dimensioni e normocollassabile con gli atti del respiro. |
La valutazione ecocardiografica (figura 2) ha evidenziato inoltre un rimodellamento concentrico del ventricolo sinistro con ipertrofia del setto interventricolare (SIV:15 mm), con una Frazione di eiezione del ventricolo sinistro normale (FE: 60 %), ridotta funzione sistolica longitudinale (S’ medio: 8.5 cm/sec), dilatazione atriale destra (RAA: 23 cmq) e aorta ascendente ectasica (44 mm).
Indagando l’anamnesi patologica familiare si è notato che un quadro simile, ma non del tutto identico, era stato già riscontrato anche nei familiari, nello specifico:
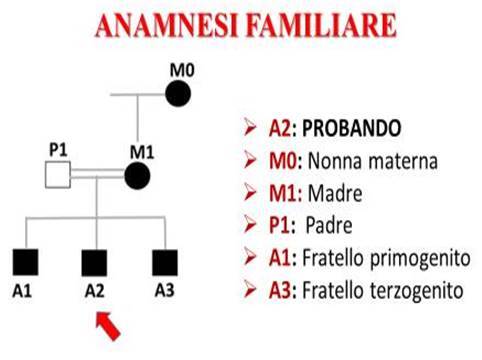
- La Nonna Materna (M0) aveva una diagnosi di Cardiomiopatia ipertrofica;
- Il Padre (P1), consanguineo di terzo grado della madre (M1), risultava apparentemente sano;
- La Madre (M1) era affetta da Cardiomiopatia ipertrofica non ostruttiva, insufficienza mitralica, ipertensione arteriosa, Tiroidite di Hashimoto, Malattia del Nodo del Seno/Sindrome Bradi-Tachi con alternanza di Flutter Atriale atipico e Bradicardia sinusale, per cui era stata sottoposta ad impianto di PM bicamerale.
- Il Fratello primogenito (A1) di 44 anni presentava un’evidenza ecocardiografica di ipertrofia del SIV (13 mm) e risultava affetto da Sindrome di Wolff-Parkinson-White (WPW) con episodi Fibrillazione Atriale Parossistica (FAP) (Figure 4 e 5);


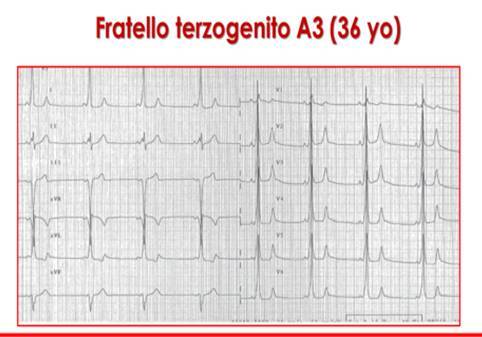
- Il Fratello terzogenito (A3) di36 annipresentava anch’egli un evidenza ecocardiografica di ipertrofia del SIV (12 mm) erisultavaaffetto da Sindrome di Wolff-Parkinson-White (WPW) con preeccitazione ventricolare da via anomala postero-settale sinistra sottoposta due volte ad ablazione con radiofrequenza (RF) (figure 6 e 7).

Alla luce del quadro clinico riscontrato nel paziente A2 e nei familiari (in particolare nei fratelli maschi), la prima ipotesi diagnostica è stata la Malattia di Danon. Essa è difatti un disordine del muscolo scheletrico e cardiaco con manifestazioni cliniche multisistemiche, X-linked dominante, associato alla mutazione a carico del gene LAMP-2 (lysosome-associated membrane protein 2) (isoforme 2°-2B-2C), implicato nei processi di autofagia cellulare del miocardio, del muscolo scheletrico e del cervello e responsabile di quadri clinici variabili, caratterizzati da miopatia a carico del muscolo cardiaco e scheletrico, disturbi del sistema di conduzione, lievi deficit intellettivi (deficit cognitivi, difficoltà nell’apprendimento), patologie retiniche, patologia epatica e patologia polmonare.
Allo scopo di confermare tale ipotesi, il probando A2 è stato sottoposto ad analisi genetica tramite Esame NGS (Next Generation Sequencing), però, contrariamente alle nostre aspettative, l’approfondimento genetico ha escluso la suddetta malattia documentando invece la presenza in eterozigosi di una variante patogenetica missenso (classe 5) del gene PRKAG2 [p.Arg302Gln (rs121908987)], correlata ad una Glicogenosi cardiaca non lisosomiale.
L’analisi genetica è stata proposta agli altri due fratelli A1 e A3 e alla madre M1, così come uno studio elettrofisiologico (SEF), al fine di valutare il substrato aritmico presente in tali pazienti, ovvero la vulnerabilità ad aritmie atriali e/o ventricolari, la capacità conduttiva della via accessoria e gli eventuali disturbi di conduzione atrio-ventricolari associati a tale sindrome, in modo da stabilire la terapia più appropriata da attuare, intesa come stretto Follow-up, ablazione della via accessoria o impianto precoce di PM o ICD/S-ICD.
Da ciò si deduce come la presenza della mutazione a carico del gene di PRKAG2 dovrebbe essere presa in considerazione in tutti i pazienti con Ipertrofia Ventricolare sinistra (LVH) e Preeccitazione ventricolare, che sviluppano Fibrillazione Atriale (FA) o necessitano di Pacemaker o Defibrillatori permanenti in giovane età.
Pertanto l’analisi genetica, insieme al dato clinico-anamnestico, svolge un ruolo cruciale poiché consente una rapida identificazione e un’appropriata gestione dei portatori genetici, non solo permettendo di fare una corretta diagnosi differenziale con altre forme di Cardiomiopatie a fenotipo ipertrofico correlate a mutazioni a carico dei geni che codificano per le proteine sarcomeriche e non, predicendo il rischio di trasmissione alla prole, ma, una volta raggiunta la diagnosi, consentendo anche una migliore stratificazione del rischio aritmico e di Morte Cardiaca improvvisa (SCD) rispetto agli Scores di rischio (HCM Risk-SCD Score) normalmente utilizzati per la cardiomiopatia ipertrofica (HCM) i quali invece tendono a sottostimare il rischio di SCD di tali pazienti, imponendo dunque un atteggiamento più aggressivo in termini di Stretto Follw-Up ed impianto precoce di ICD, s-ICD e PM.
BIBLIOGRAFIA.
1.Lev M, Leffler WB, Langendorf R, Pick A. Anatomic findings in a case of ventricular preexcitation (WPW) terminating in complete atrioventricular block. Circulation 1966;34: 718–33.
2.MacRae CA, Ghaisas N, Kass S, et al. Familial hypertrophic cardiomyopathy with WolffParkinson-White syndrome maps to a locus on chromosome 7q3. J Clin Invest 1995;96:1216–20.
3.Gollob MH, Green MS, Tang AS, et al. Identification of a gene responsible for familial WolffParkinson-Whitesyndrome. N Engl J Med 2001; 344:1823–31.
4.Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol 2016;68:2871–86.
5.Maron BJ, Roberts WC, Arad M, et al. Clinicaloutcome and phenotypic expression in LAMP2cardiomyopathy. JAMA 2009;301:1253–9.
6.Lopez-Sainz A, Dominguez F, Lopes LR, et al. Clinical Features and Natural History of PRKAG2 Variant Cardiac Glycogenosis J Am Coll Cardiol. 2020;76:186-197. Danon disease: clinical features, evaluation, and management.
7.D’souza RS, Levandowski C, Slavov D, et al Circ Heart Fail. Danon disease: clinical features, evaluation, and management. 2014;7:843-9
