
“Se senti zoccoli…questa volta pensa alle zebre e non ai cavalli”
Una donna di 50 anni, affetta da drepanocitosi e sottoposta a recente vaccinazione anti SARS-CoV2 (III° dose) giunge in Pronto Soccorso accusando artromialgie diffuse, localizzate in particolare agli arti inferiori. Viene posta diagnosi di crisi drepanocitica, probabilmente scatenata dalla vaccinazione, e la donna viene ricoverata per proseguire le cure del caso.
Dopo circa cinque giorni dal ricovero, la paziente lamenta dispnea ingravescente associata a dolore toracico. Si esegue un elettrocardiogramma che mostra: ritmo sinusale a frequenza di 91/m’, normale conduzione AV. Pattern S1 Q3 T3 ed onde T negative a sede precordiale. (Fig. 1)
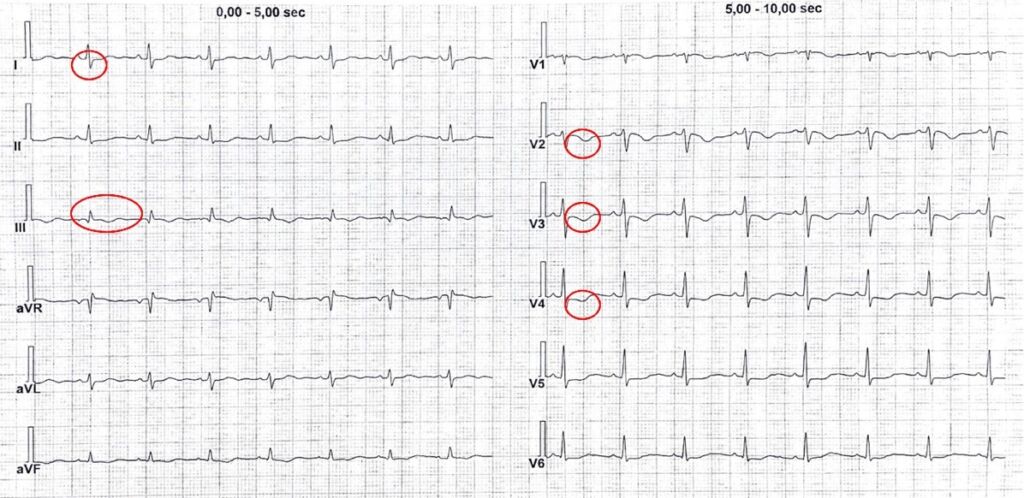
Figura 1: ECG
L’elettrocardiogramma effettuato al ricovero non mostrava le alterazioni da sovraccarico destro appena rilevate (Fig. 2).
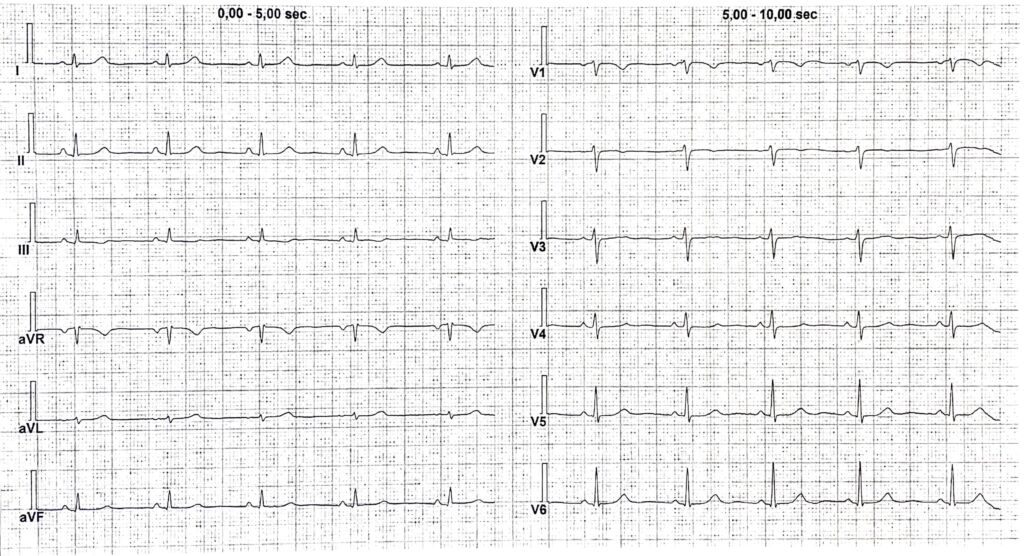
Figura 2: ECG basale
Viene inoltre effettuato un ecocardiogramma che mostra il ventricolo destro dilatato (RVOT PLAX 35 mm; RVD1 42 mm; RVD2 32 mm) con funzione sistolica longitudinale lievemente ridotta (TAPSE 17 mm), D-Shape del setto interventricolare con eucinesia dei restanti segmenti e funzione globale di pompa conservata (FE 60%). Atrio destro anch’esso dilatato (RA 21,5 cmq). Insufficienza tricuspidalica moderata-severa con elevati valori di PAPs stimata (65-70 mmHg). Vena cava inferiore dilatata e scarsamente collassabile con gli atti del respiro. (Fig. 3 e 4)

Figura 3: D-Shape SIV
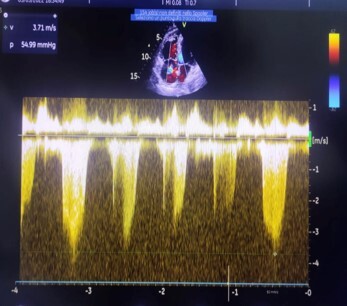
Figura 4: PAPs
Con questi elementi clinico-strumentali, nel forte sospetto di un’embolia polmonare acuta, viene effettuata un Angio-TC del torace, che però risulta negativa per minus attribuibili alla presenza di apposizioni trombotiche a carico dei principali rami in esame bilateralmente.
Cosa può aver causato un’elevazione acuta della pressione arteriosa polmonare tale da causare segni clinici, ECGrafici ed ecocardiografici di sovraccarico ventricolare destro?
Discussione:
La Drepanocitosi (o Anemia Falciforme) è una delle più comuni emoglobinopatie su base genetica. È causata da una mutazione puntiforme nel gene che codifica per la catena beta dell’emoglobina e che determina la sostituzione dell’acido glutammico (un amminoacido idrofilico) con la valina (un amminoacido idrofobico). Questo abbassa la solubilità della proteina in configurazione deossigenata e determina la precipitazione dell’emoglobina con formazione di fibrille nei globuli rossi e la loro caratteristica forma “a falce”(Fig. 5). La falcizzazione degli eritrociti non è costitutiva ma si determina in particolari condizioni (ipossia, acidosi, innalzamento della temperatura, ecc.).

Figura 5: Striscio di sangue di un paziente affetto da anemia falciforme
Inoltre, circa il 39% dei pazienti affetti, sperimenta nel corso della sua vita diverse crisi falcemiche vaso-occlusive associate a dolore (VOC), che interessano torace, addome e articolazioni: eventi gravi, imprevedibili, che possono rappresentare delle vere emergenze sanitarie. Indagando la letteratura, è stato recentemente documentato durante le crisi vaso-occlusive e durante l’esercizio fisico un’elevazione in acuto della pressione polmonare.[1]
Questa presentazione clinica, è spesso accompagnata da severa anemia, emolisi e vasocostrizione.[2]
Il meccanismo fisiopatologico che porta all’instaurarsi dell’ipertensione polmonare nell’anemia falciforme, come anche in altri disordini emolitici cronici, è complesso. Il comune denominatore alla base dello sviluppo dell’ipertensione polmonare è probabilmente costituito dall’emolisi cronica. Diversi studi, infatti, mostrano una forte associazione tra la severità dell’emolisi e lo sviluppo di ipertensione polmonare[3;4].

Figura 6: Fisiopatologia dell’ipertensione polmonare nei disordini emolitici
L’emoglobina libera rilasciata durante le crisi emolitiche spiazza il vasodilatatore intrinseco rappresentato dall’ossido nitrico (NO); inoltre la rottura dei globuli rossi rilascia l’arginasi, un enzima responsabile della deplezione dell’L-arginina, substrato chiave per la sintesi dell’NO. L’ossido nitrico è uno dei più potenti vasodilatatori attualmente conosciuti e ha un ruolo essenziale nell’omeostasi vascolare: è fondamentale nel mantenere il tono vasomotore, nel limitare l’aggregazione piastrinica e il danno da ischemia-riperfusione, modula la proliferazione endoteliale e ha anche proprietà anti-infiammatorie[5].
L’inattivazione e la ridotta sintesi dell’NO porta ad uno squilibrio della vasodilatazione a livello dei vasi polmonari. L’arginasi è anche responsabile dell’alterato metabolismo dell’L-arginina in L-ornitina che porta alla sintesi in ultima istanza di L-prolina, la quale contribuisce alla proliferazione delle cellule muscolari lisce e alla sintesi del collagene, risultando in un rimodellamento vascolare ed ispessimento intimale[6].
Le strategie di trattamento di questa complicanza includono i rimedi specifici dei disordini emolitici, come trasfusioni di sangue, chelanti del ferro, idrossiurea e l’ossigeno-terapia, che possono prevenire lo sviluppo e la progressione di ipertensione polmonare.
Il ruolo di terapie specifiche per l’ipertensione polmonare non è stato ancora stabilito[7].
Bibliografia:
1 Machado RF, Mack AK, Martyr S, Barnett C, Macarthur P, Sachdev V, et al. Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol 2007;136:319-25.
2 Lechapt E, Habibi A, Bachir D, Galacteros F, Schaeffer A, Desvaux D, Brochard L, Housset B, Godeau B, Maitre B. Induced sputum versus bronchoalveolar lavage during acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med 2003;168:1373–1377.
3 Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350:886-95.
4 Kato GJ, Onyekwere OC, Gladwin MT. Pulmonary hypertension in sickle cell disease: Relevance to children. Pediatr Hematol Oncol 2007;24:159-70.
5 Ataga KI, Moore CG, Jones S, Olajide O, Strayhorn D, Hinderliter A, et al. Pulmonary hypertension in patients with sickle cell disease: A longitudinal study. Br J Haematol 2006;134:109-15.
6 Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 2007;110:2166-72.
7 Morris CR, Vichinsky EP, van Warmerdam J, Machado L, Kepka- Lenhart D, Morris SM Jr, et al. Hydroxyurea and arginine therapy: Impact on nitric oxide production in sickle cell disease. J Pediatr Hematol Oncol 2003;25:629-34.
