
Don’t go breaking my heart
Caso clinico:
Un uomo di 53 anni, affetto da ipertensione arteriosa sistemica e diabete mellito di tipo II in terapia con ipoglicemizzanti orali, in marzo 2021 allertava il 118 per insorgenza improvvisa di malessere diffuso associato a nausea e vomito. All’arrivo dei soccorritori il paziente si presentava emodinamicamente stabile ma tachicardico e tachipnoico: la pressione arteriosa registrata era di 120/80 mmHg, la frequenza cardiaca di 115 battiti/minuti e la frequenza respiratoria di 30 atti/minuti.

Fig. 1 Elettrocardiogramma sul territorio: tachicardia sinusale, FC 115 bpm. Sovraslivellamento del tratto ST in sede anterolaterale.
All’elettrocardiogramma (Figura 1) si registrava una tachicardia sinusale con sopraslivellamento del tratto ST in sede antero-laterale: nel sospetto di un infarto miocardico acuto tipo STEMI il paziente veniva trasportato in urgenza in sala di Emodinamica del nostro Policlinico. Le coronarie sono risultate indenni da lesioni angiograficamente indenni (Figura 2), mentre alla ventricolografia l’aspetto era suggestivo per sindrome di Tako-tsubo con ipercontrattilità dei segmenti basali del ventricolo sinistro e un apice cardiaco acinetico (Figura 3). Durante la procedura veniva posizionato un contropulsatore aortica per insorgenza di severa ipotensione.

Fig. 2 Coronarografia: arterie coronarie indenni da lesioni angiograficamente significative.
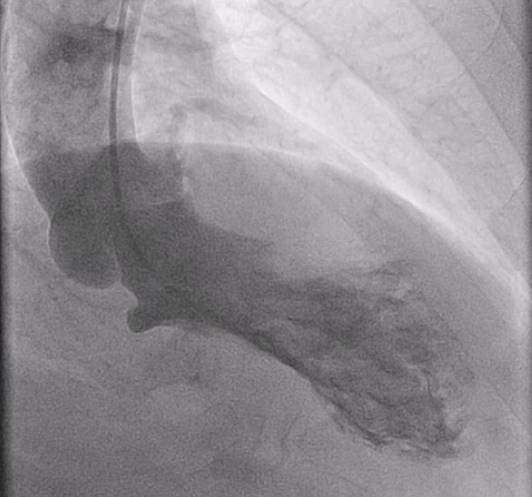
Fig. 3 Ventricolografia: ipercontrattilità dei segmenti basali con acinesia dell’apice.
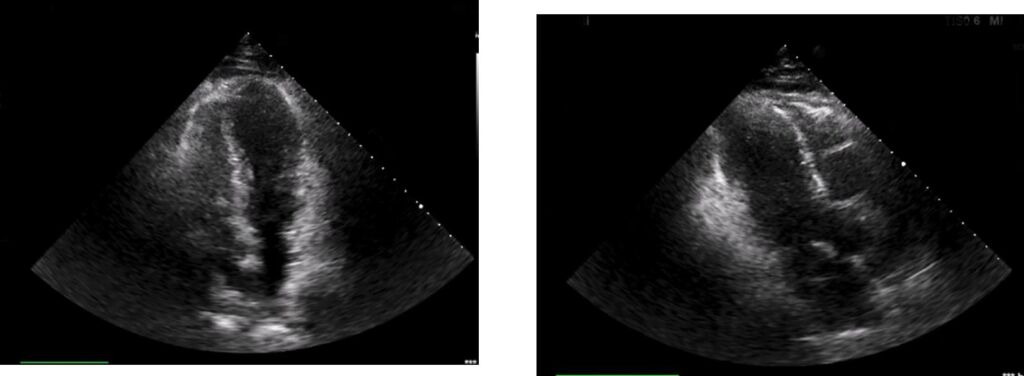
Il paziente veniva pertanto trasferito in Terapia Intensiva Cardiologica: all’elettrocardiogramma (Figura 4) si osservava l’evoluzione a onde T negative/difasiche diffuse con contestuale allugamento del tratto QT; l’ecocardiogramma (Figura 5) confermava il quadro ventricolografico, con una funzione sistolica globale severamente depressa (Frazione di eiezione stimata circa 30%). Agli esami di laboratorio si osservava un leucocitosi neutrofila, un danno renale acuto che si sarebbe successivamente risolto nel corso della degenza e un quadro di diabete mellito scompensato.
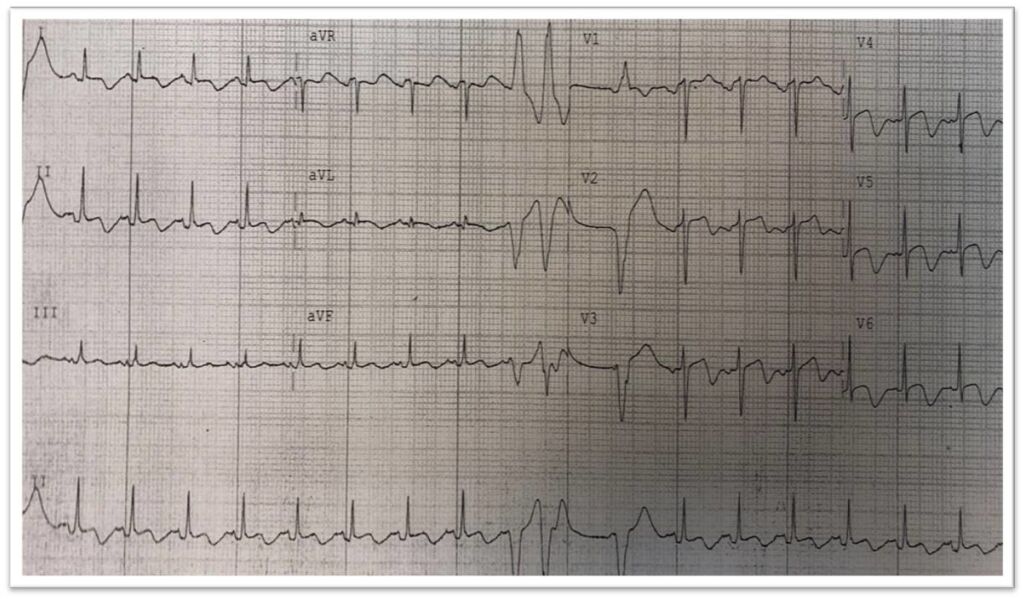
Fig. 4 Elettrocardiogramma all’ingresso in Terapia Intensiva: evoluzione a onde T negative/bifasiche con allungato dell’intervallo QT.

Fig. 5 Ecocardiogramma: funzione sistolica del ventricolo sinistro severamente depressa con frazione di eiezione uguale a 30%. Acinesia di tutti i segmenti apicali con ipercontrattilità dei segmenti basali.
Raccogliendo l’anamnesi, si è venuti a conoscenza che il paziente era già stato ricoverato in giovane età per un episodio perimiocarditico e per un infarto miocardico acuto tipo NSTEMI a coronarie angiograficamente indenni nel 2018, dove contestualmente erano state escluse tramite AngioTc embolia polmonare e sindrome aortica acuta; allora veniva segnalato un incidentaloma surrenalico di 59 mm che non sarebbe stato successivamente indagato in maniera mirata. Nel 2018 la risonanza magnetica eseguita alla dimissione mostrava una normale funzione sisto-diastolica biventricolare e una sfumata area di Late Gadolinium Enhancement (LGE) compatibile con pregresso episodio miocarditico.
Tornando al 2021, il giorno successivo al ricovero è stato necessario rimuovere il contropulsatore aortico per insorgenza di severa ipertensione arteriosa sistemica, refrattaria successivamente anche alla somministrazione di nitrato ev. Veniva pertanto eseguita una Tc encefalo per escludere emorragia cerebrale, risultata negativa, e una AngioTC dell’aorta toracoaddominale, che escludeva nuovamente una sindrome aortica acuta. Come reperto accessorio, la Tc mostrava nelle scansioni addominali un netto incremento dimensionale della nota lesione surrenalica sinistra (73×57 mm vs 59×44 mm, rispetto al 2018), non captante contrasto (compatibile quindi con lesione surrenalica non adenomatosa, Figura 6). Nel sospetto di una forma secondaria di ipertensione arteriosa, sono stati eseguiti i dosaggi dei più comuni ormoni, risultando in livelli particolarmente elevati di metanefrine plasmatiche (6300 pmol/L, valori normali < 45 pg/L) e urinarie (2148 microg/die, valori normali 64-302 microg/die).
Il sospetto di feocromocitoma surrenalico secernente venne confermato tramite l’esecuzione di una DOTANOC- PET con tracciante marcato con 8-Ga: l’esame mostrò un marcato uptake del tracciante (SUV 12,9) nella regione più craniale e mediale del surrene sinistro, compatibile con tumore neuroendocrino con espressione di recettori per somatostatina.
Dunque, è stata intrapresa terapia profilattica prima con alfalitici e in secondo luogo con betabloccanti in preparazione della chirurgia e successivamente il paziente è stato sottoposto a una surrenectomia sinistra per via laparoscopica.
L’elettrocardiogramma e l’ecocardiogramma eseguiti nel corso della degenza sono andati in contro a progressivo miglioramento delle anomalie prima descritte fino a pressoché completa normalizzazione.
La risonanza magnetica pre-dimissione ha confermato la normalizzazione della funzione sistolica del ventricolo sinistro con invariata la stria di LGE anterolaterale compatibile con il pregresso episodio miocarditico (Figura 7).

Fig. 7 Risonanza magnetica cardiaca: normalizzazione della funzione contrattile del ventricolo sinistro.
Cosa ci dice la letteratura
Il feocromocitoma è un tumore endocrino che origina da cellule catecolaminergiche della midollare del surrene; ha un’incidenza di 3-5 casi su 100.000 nella popolazione generale e viene riscontrato con diagnosi accidentale nel 10-30% dei casi1. Nel 30-60% dei pazienti è possibile riscontrare una mutazione genetica alla base della neoplasia (nelle sindromi da neoplasie endocrine multiple 2 da mutazione di RET, nella sindrome di Von Hippel Lindau da mutazione del gene VHL e nella neurofibromatosi/malattia di von Recklinghausen da mutazione di NF1)2.. Le manifestazioni cliniche sono determinate dalla increzione nel torrente ematico di catecolamine che determinano importanti ripercussioni cardiocircolatorie.
Il quadro clinico è estremamente pleomorfo: la manifestazione più frequente è costituita dall’ipertensione arteriosa sistemica, di cui il feocromitoma è una causa da prendere in considerazione nelle forme secondarie (in cronico o parossistica, come crisi ipertensiva); si può manifestare inoltre con aritmie, a volte anche mortali (come tachicardia o fibrillazione ventricolare) o con emergenze cliniche come shock cardiogeno, infarto miocardico o dissezione aortica.
Le ripercussioni dell’iperincrezione in circolo di catecolamine a livello cardiaco si configurano nel quadro di cardiomiopatia catecolaminergica, che si osserva nell’8-11% dei pazienti con feocromocitoma: seppur una complicanza rara, essa complica la prognosi del paziente con feocromocitoma e aumenta il rischio chirurgico. Questa si può manifestare come un quadro “simil-cardiomiopatia ipertrofica” che può sfociare nella sua forma dilatativa, e come sindrome di Takotsubo3.. Questa è determinata dall’eccessiva secrezione in circolo, ad andamento parossistico, di catecolamine che, a livello cardiaco, andando a stimolare in maniera sproporzionata i recettori beta-adrenergici determinano vasospasmo e danno miocellulare con distrettualità della cinetica diverse a seconda della diversa distribuzione delle fibre nervose simpatiche nei pazienti, cosa che sfocia nelle manifestazioni classiche della sindrome di Tako-tsubo (apicale, basale o reverse, medio-ventricolare, focale) 4.. Le forme di sindrome di Tako-tsubo secondarie a feocromocitoma hanno caratteristiche diverse rispetto a quelle classiche: il “gender gap” è meno sproporzionato verso il sesso femminile (colpito nel 70% nelle forme da feocromocitoma, 89% nelle forme classiche); sono più rappresentate le forme al di fuori della apicale (la più frequente nella forma classica), la frazione di eiezione è significativamente inferiore all’ingresso e si complicano più frequentemente con shock cardiogeno durante la degenza, ma la mortalità e la prognosi sono significativamente migliori rispetto alla forma classica, a sottolineare la presenza di una causa correggibile alla base della sindrome 4.. Questo spiega come mai in passato (e ancora oggi secondo gli Autori Giapponesi) il feocromocitoma venisse considerato criterio di esclusione per la sindrome di Takotsubo: di contro oggi, secondo il Consensus Document della Società Europea di Cardiologia pubblicato nel 2018 questa neoplasia viene considerata come possibile stressor in questa sindrome rientrando di fatto nei suoi criteri diagnostici. 5
In merito alla terapia del feocromocitoma, l’unica curativa rimane la rimozione chirurgica della neoplasia: essa deve essere condotta con la minor manipolazione possibile della massa (“No touch technique”) in quanto essa può determinare crisi ipertensive tramite l’immissione in circolo di catecolamine; per minimizzare i rischi deve essere intrapresa almeno dieci giorni prima della chirurgia terapia con alfa-bloccanti e, solo successivamente, con betabloccanti, in quanto la loro somministrazione in assenza di alfalitici può determinare crisi ipertensive (per iperstimolazione incontrastata da parte delle catecolamine dei recettori alfa) 6,7.
Bibliografia
- Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS OF PARAGANGLIOMA: Clinical manifestations of paraganglioma syndromes types 1-5. Endocr Relat Cancer. 2015;22:T91-103.
- Mercado-Asis LB, Wolf KI, Jochmanova I, Taïeb D. PHEOCHROMOCYTOMA: A GENETIC AND DIAGNOSTIC UPDATE. Endocr Pract. 2018;24:78-90.
- Santos JRU, Brofferio A, Viana B, Pacak K. Catecholamine-Induced Cardiomyopathy in Pheochromocytoma: How to Manage a Rare Complication in a Rare Disease? Horm Metab Res. 2019; 51:458-469.
- Y-Hassan S, Falhammar H. Pheochromocytoma- and paraganglioma-triggered Takotsubo syndrome. Endocrine. 2019; 65(3):483-493.
- Ghadri JR, Wittstein IS, Prasad A, Sharkey S, Dote K, et al. International Expert Consensus Document on Takotsubo Syndrome (Part I): Clinical Characteristics, Diagnostic Criteria, and Pathophysiology. Eur Heart J. 2018 ;39:2032-2046.
- Galati SJ, Said M, Gospin R, Babic N, Brown K, et al. The Mount Sinai clinical pathway for the management of pheochromocytoma. Endocr Pract. 2015 ;21:368-82.
- Pappachan JM, Tun NN, Arunagirinathan G, Sodi R, Hanna FWF. Pheochromocytomas and Hypertension. Curr Hypertens Rep. 2018;20:3.
