Nell’Aprile 2019 un uomo di 36 anni, iperteso, fumatore, dislipidemico, giungeva alla nostra attenzione in regime di consulenza con richiesta di nullaosta a terapia oncologica con Ifosfamide per sarcoma sinoviale mediastinico.
Il paziente, nonostante la giovane età, riferiva una storia cardiologica complessa: tre pregressi episodi di infarto miocardico acuto esorditi con dolore toracico e rialzo degli indici di miocardionecrosi a seguito dei quali veniva eseguito studio coronarografico che mostrava coronarie epicardiche indenni da lesioni (rispettivamente nel 1997, 2004 e 2011); un episodio di fibrillazione atriale ad elevata risposta ventricolare cardiovertita con amiodarone (2006) ed un episodio di tachicardia ventricolare sostenuta a seguito della quale veniva impiantato un defibrillatore in prevenzione secondaria (2018). Inoltre nel 2011 aveva eseguito una RMN cardiaca, con riscontro di disfunzione ventricolare destra associata a necrosi del setto infero-apicale del ventricolo sinistro.
Nel febbraio 2019 per insorgenza di dispnea ingravescente e riscontro di versamento pleurico massivo alla radiografia, il paziente si sottoponeva a toracentesi ed esame istologico/citologico del liquido sinoviale che poneva diagnosi di sarcoma sinoviale mediastinico. Veniva contestualmente eseguita TC del torace per migliore caratterizzazione della lesione mediastinica che evidenziava la presenza “multipli gettoni di tessuto patologico a base pleurica con estrinsecazione a livello mediastinico, in particolare si descriveva quota tissutale in sede carenale/sottocarenale paramediana destra con DT 9 cm” (Figura 1).
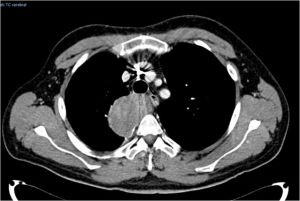
Al momento della visita il paziente appariva in mediocri condizioni cliniche generali (PA 110/70, FC 70 bpm, SO2 97% con VMK 31%), obiettività cardiaca nella norma ed evidenza all’esame obiettivo polmonare di crepitii diffusi con murmure vescicolare ridotto a livello basale destro. All’ECG evidenza di ritmo sinusale interrotto da extrasistoli ventricolari, blocco di branca destra, onde T invertite da V1 a V4 (Figura 2). Agli esami ematici evidenza di ipo-natriemia (Na 128 mmol/l), rialzo degli indici di danno epatico e di colestasi (AST-GOT 85 U/l, ALT-GPT 234 U/L, gamma-GT 561 U/l, fosfatasi alcalina 148 U/l, LDH 303 U/l) e dell’NT-proBNP (988 pg/ml).

Si procedeva quindi ad esecuzione di ecocardiogramma transtoracico che mostrava un “ventricolo destro dilatato con diffusa iper-trabecolatura, globale ipocinesia e marcata disfunzione (FAC 16%); ventricolo sinistro di dimensioni ai limiti alti della norma con lieve ipertrofia parietale e diffusa ipocinesia (FE 30%)” (Figura 3).

Riesaminando i dati raccolti emergeva come possibile diagnosi quella di displasia aritmogena del ventricolo destro. Secondo i criteri diagnostici stabiliti dalla Task Force Internazionale del 2010, erano già presenti un criterio maggiore (evidenza ecocardiografica di > 1 aree ipocinetiche con frazione di accorciamento del ventricolo destro < 33%) e due criteri minori (pregressa tachicardia ventricolare sostenuta ad asse sconosciuto, evidenza elettrocardiografica di onde T negative da V1 a V4). (1)
Veniva inoltre presa in esame la TC torace precedentemente eseguita con particolare attenzione all’immagine cardiaca che mostrava dilatazione biventricolare ed aspetto assottigliato/trabecolato della parete libera del ventricolo destro (Figura 4).

Si propendeva quindi ad esecuzione di RMN cuore a conferma del sospetto clinico. Quest’ultima metteva in evidenza diffusa ipocinesia ventricolare destra, assottigliamento di parete ed aneurisma focale a livello del tratto di efflusso del ventricolo destro con potenziamento contrastografico tardivo a livello della parete libera (soprattutto versante medio-basale), tale reperto era evidente anche a livello della parete infero-posteriore e laterale del ventricolo sinistro che risultava inoltre ipocontrattile (FE 35%). Si poneva diagnosi di Displasia aritmogena del ventricolo destro con disfunzione cardiaca biventricolare; veniva dato nullaosta al trattamento del sarcoma sinoviale mediastinico seppur con rischio cardiologico elevato.
COMMENTO
La displasia aritmogena del ventricolo destro è una malattia che colpisce il miocardio ed ha alla base difetti genetici con mutazione delle proteine desmosomiali quali placoglobina, desmoplachina, placofillina, desmogleina e desmocollina.(2-4) Tali mutazioni inducono ad una sostituzione del muscolo cardiaco con tessuto fibro-adiposo, progressiva disfunzione ventricolare ed insorgenza di aritmie (tachicardia ventricolare e fibrillazione ventricolare) che possono portare a morte improvvisa.(3)
La classificazione attualmente in uso include tre varianti: 1) la forma classica con coinvolgimento del ventricolo destro 2) la forma biventricolare 3) la forma con coinvolgimento isolato del ventricolo sinistro.(5-7)
La storia naturale della forma classica prevede iniziale coinvolgimento del ventricolo destro ma nelle fasi più avanzate può coinvolgere anche il ventricolo sinistro fino al quadro di “disfunzione cardiaca biventricolare”. (2, 4, 8) Esaminando il caso clinico emerge come il paziente sia già ad uno stadio di “displasia aritmogena del ventricolo destro con disfunzione cardiaca biventricolare”.
Anche la storia clinica del paziente in esame ripercorre le fasi tipiche della patologia, con iniziali episodi di dolore toracico, anomalie della ripolarizzazione all’ECG e rialzo degli indici di miocardionecrosi ma coronarie epicardiche indenni da lesioni (insorti nel 1997, 2004 e 2011). (9) In questa fase la displasia aritmogena entra in diagnosi differenziale con la miocardite che però veniva esclusa per assenza di reperti clinici (incremento degli indici di flogosi e rialzo termico) ed ecocardiografici suggestivi (ipocinesia in sede infero-laterale del ventricolo sinistro con versamento pericardico, iperecogenicità miocardica, recupero rapido della contrattilità ventricolare).(10)
In una fase più avanzata predominano gli episodi di Tachicardia o Fibrillazione ventricolare, evento coincidente con la formazione di tessuto cicatriziale fibro-adiposo (nel caso del paziente insorto nel 2018). Infine, subentra il quadro di disfunzione del ventricolo destro e successivamente biventricolare, stadio in cui il paziente si presentava al momento della consulenza (2019).(9)
Dal punto di vista terapeutico il primo obiettivo è la prevenzione della morte cardiaca improvvisa, il paziente infatti era già portatore di ICD impiantato precedentemente in prevenzione secondaria. È inoltre importante l’ottimizzazione della terapia della disfunzione ventricolare: beta bloccante e sartano erano già in terapia, è stata aggiunta terapia con antagonista dell’aldosterone. (11)
BIBLIOGRAFIA
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121(13):1533-41.
- Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet (London, England). 2009;373(9671):1289-300.
- Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circulation Cardiovascular genetics. 2013;6(6):533-42.
- Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart (British Cardiac Society). 2011;97(7):530-9.
- Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circulation research. 2017;121(7):784-802.
- Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115(13):1710-20.
- Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. Journal of the American College of Cardiology. 2008;52(25):2175-87.
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. The New England journal of medicine. 1988;318(3):129-33.
- Lemola K, Brunckhorst C, Helfenstein U, Oechslin E, Jenni R, Duru F. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: long term experience of a tertiary care centre. Heart (British Cardiac Society). 2005;91(9):1167-72.
- Ammirati E, Frigerio M, Adler ED, Basso C, Birnie DH, Brambatti M, et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circulation Heart failure. 2020;13(11): e007405.
- Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41(14):1414-29.